Because half of all instances of hearing loss are linked to genetic mutations, advanced gene research is an invaluable tool for uncovering causes of deafness—and one of the biggest hopes for the development of new therapies. Now Professor Karen Avraham of the Sackler Faculty of Medicine at Tel Aviv University has discovered a significant mutation in a LINC family protein—part of the cells of the inner ear—that could lead to new treatments for hearing disorders.
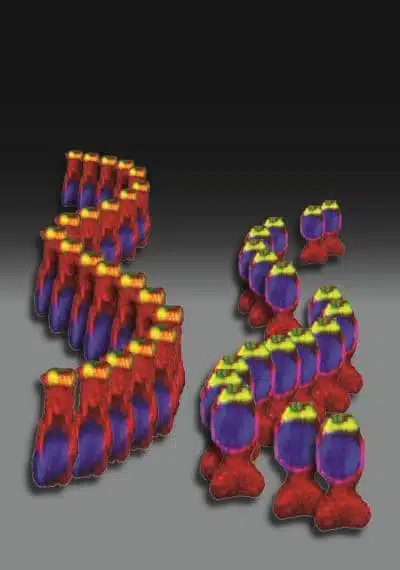
|
|
The different position of cell nuclei in |
Her team of researchers, including Dr Henning Horn and Professors Colin Stewart and Brian Burke of the Institute of Medical Biology at A*STAR in Singapore, discovered that the mutation causes chaos in a cell’s anatomy. The cell nucleus, which contains an individual’s entire DNA, moves to the top of the cell rather than being anchored to the bottom, its normal place. Though this has little impact on the functioning of most of the body’s cells, it’s devastating for the cells responsible for hearing, explains Avraham. “The position of the nucleus is important for receiving the electrical signals that determine proper hearing,” she explains. “Without the ability to receive these signals correctly, the entire cascade of hearing fails.”
This discovery, recently reported in the Journal of Clinical Investigation, may be a starting point for the development of new therapies. In the meantime, the research could lead toward work on a drug that is able to mimic the mutated protein’s anchoring function, and restore hearing in some cases, she suggests.
From human to lab to mouse. Avraham originally uncovered the genetic mutation while attempting to explain the cause of deafness in two families of Iraqi Jewish descent. For generations, members of these families had been suffering from hearing loss, but the medical cause remained a mystery. Using deep genetic sequencing, a technology used to sequence the entire human genome, she discovered that the hearing impaired members of both families had a mutated version of the protein Nesprin4, a part of the LINC group of proteins that links the cell’s nucleus to the inner wall of the cell.
In the lab, Avraham recreated this phenomenon by engineering the mutation in single cells. With the mutation in place, Nesprin4 was not found in the area around the cell nucleus, as in healthy cells, but was spread throughout the entire cell. Investigating further, she studied lab mice that were engineered to be completely devoid of the protein.
Created in Singapore, the mice were originally engineered to study the biology of LINC proteins. The fact that they were deaf came as a complete surprise to researchers. Without this protein serving as an anchor, the cell nucleus is not located in the correct position within inner ear cells, but seems to float throughout. This causes the cells’ other components to reorient as well, ultimately harming the polarity of the cells and hindering electrical signals. The mutation took a heavy toll on the cells’ ability to transfer sound signals, explains Avraham, rendering the mice deaf.
Given the similarity between mouse and human inner ear cells, researchers predict that the same phenomenon is occurring in human patients with a mutation in the Nesprin4 gene.
Looking for a wider impact. Avraham says that she and her collaborators are the first to reveal this mutation as a cause of deafness. “Now that we have reported it, scientists around the world can test for mutations in this gene,” she notes. The mutation could indeed be a more common genetic cause of deafness in a number of populations. Because Nesprin4 belongs to a family of proteins that have been linked to other diseases, such as muscular coordination and degeneration disorders, this could prove a ripe area for further research.
At TAU, the research was supported by the National Institutes of Health—NIDCD and Israeli Center of Research Excellence, I-CORE.
Source: American Friends of Tel Aviv University